Why BitQT ?¶
There are plenty of clustering algorithms for analyzing MD out there, so why QT over those others? Well, not all kind of algorithms suit all problemic situations with the same performance. In those particular cases where strongly geometrically correlated conformations are needed to be returned as clusters, QT stands out as an ideal option because that guarantees that no pair of frames having a similarity value greater than a user-specified cutoff will coalesce into the same cluster.
This sounds great, but in practice, exact (and even approximate) implementations of QT are computationally expensive. That’s where BitQT enters the scne. This heuristic makes a parallel with the Maximum Clique Problem (MCP) and treats QT problem as a search of cliques in a mathemathical graph. As it is possible to conduct this search using vector of bits, BitQT is a fast and memory efficient alternative to the few other current implementations.
How Does BitQT Works ?¶
Here we expose briefly the basis of BitQT assumptions and the key aspects of the algorithm. You can refer to the academic publication for more details.
Original QT algorithm¶
The application of the original QT to an MD trajectory can be described as follows: After the selection of a similarity threshold k, one arbitrary frame is selected and marked as a candidate cluster C1. The remaining frames are iteratively added to C1 if and only if two conditions hold;
Condition 1: the similarity distance between the entering frame and every frame inside C1 is the minimum possible, and
Condition 2:- the similarity distance between the entering frame and every frame already inside C1 does not exceed the threshold k.
This process continues for all frames n in the trajectory until Cn candidate clusters have been formed. The one with more frames is set as a cluster, its elements removed from further consideration, and the entire process repeated until no more clusters can be discovered.
Parallel with the Maximum Clique Problem¶
The most importat aspect of the original algorithm is its guarantee that all pairwise similarities inside a cluster will remain under the threshold k. This aspect is assured by Condition 2. Condition 1 merely limits the size of retrieved clusters but has no impact in maintaining the similarity threshold.
From Graph Theory, we know that a clique is a subgraph in which vertices are all pairwise adjacent. If a clique is not contained in any other clique, it is said to be maximal, while the term maximum clique denotes the maximal clique with a maximum number of nodes. The maximum clique problem (MCP) deals with the challenge of finding the maximum clique inside a given graph.
To make a parallel between QT and the MCP, we represent each frame of an MD trajectory as a node of an undirected graph in which edges depict RMSD similarity between nodes. Only edges with an RMSD less or equal than the threshold k are allowed. In that context, QT can be declared as an iterative search of cliques. QT cliques, however, are not necessarily maximum due to Condition 1 of the algorithm, which ensures that they should have a minimum weight instead of a maximum cardinality.
Conveniently, a redefinition of the QT algorithm can be made to search for maximum-sized clusters instead of minimum-weighted without compromising the pairwise similarity assured by the Condition 2. Relaxation of Condition 1 in this way, automatically converts QT in an MCP problem, accessible by the graph theory tools.
This approach has a profound impact on how molecular similarity can be encoded and in the efficiency of algorithms that can be used to solve the problem, as discussed in the next sections.
BitQT Algorithm¶
1. RMSD-encoded Binary Matrix
If we conceive the QT algorithm as an MCP problem, after relaxation of Condition 1 our search will be focused on finding cliques of maximum cardinality, and no useful information is extracted from the weight of the edges other than its absence or existence. This information can therefore be encoded as a binary matrix M where M_ij=1 if nodes i and j are similar (RMSD_ij <= k) or 0 otherwise.
Besides the RAM saving, expressing similarity as a binary matrix offers the possibility to perform the search of cliques using binary operators (AND and XOR), contributing to the speedup of the heuristic we propose in the following sections.
2. Nodes coloring
Each vertex of the input graph (Graph 1, Figure 1) is ranked (column R, Matrix 1, Figure 1) in descending order of their corresponding degrees (column D, Matrix 1, Figure 1). Following the rank order, each vertex takes a color label that it shares with all other vertices that are neither colored nor neighbors (column C, Matrix 1, Figure 1).

Figure 1: Workflow diagram of BitQT algorithm¶
3. Clique search from the maximum degree node
After all vertices are colored, the search of a clique starts considering only neighbors of the maximum degree node of the graph (Graph 1A, Figure 1), which is called the seed of the clique (node 1 in Matrix 1A, Graph 1A, Figure 1). Neighbors of the seed are strictly ordered for further processing following three criteria (DCg ordering); descending order of their degrees, ascending order of their color class, and ascending order of the degeneracy of the color class (columns D, C, and g, respectively, Matrix 1A, Figure 1). Following this ordering, the first node is selected to start a clique, and subsequent nodes will be added to that clique if they have a still-not-explored color and if they are adjacent to previously explored nodes (clique propagation).
BitQT performs this search using bitwise operations. The bit-vector Bi corresponding to the maximum degree node is set as the clique bit-vector (B1 in Heuristic search of Graph 1A, Figure 1). Following the DCg ordering, an AND operation is performed between the clique bit-vector and the next node bit-vector if it has a new color (B6 in Heuristic search of Graph 1A, Figure 1). Indices corresponding to bits that become zero by this operation are discarded from further consideration (B2, B3, B4, and B5) as they are not adjacent to processed nodes (B1 and B6). The resulting bit-vector becomes the new clique bit-vector used for the AND operation with the next candidate following the DCg ordering (B9). The bit-vector resulting from the iterative AND operations contains the members of the first clique.
4. Clique search from promising nodes
Once the clique retrieved by using the maximum degree node as the seed is found in the previous step, the same exploration strategy is conducted for every emph{promising node} in the original graph (Graph 1). A promising node (B8 in Graph 1, Figure 1) is defined as a node with a color not present in the first clique and whose degree is higher than the number of nodes in the first clique. Using such nodes as seeds for propagation might lead to the formation of a bigger clique (Heuristic search of Graph 1B, Scheme 1).
5. Conclusion and updating
When the maximum degree node and all promising nodes have been used as seeds, the maximum clique found is picked as a cluster, and their nodes removed from the input graph (the corresponding Bi vectors removed from the binary matrix). An updating of the remaining bit-vector is necessary to set as zero all entries corresponding to nodes that formed the cluster, which will not be available for subsequent iterations. This updating is bitwise encoded as a consecutive AND/XOR operation between remaining bit-vectors and the clique bit-vector (Conclusion of iteration 1, Figure 1). The same steps are repeated from Step 3 until no more cliques can be found.
Performance Benchmark¶
The two QT implementations used for comparisons correspond to the QT code (QTPy), and the qtcluster command distributed in version 6.0.1 of the ORAC package.
MD trajectories of different sizes and compositions were selected: 6K- a 6001 frames REMD simulation of the Tau peptide, 30K- a 30605 frames MD of villin headpiece based on PDB 2RJY, 50K- a 50500 frames MD of serotype 18C of Streptococcus Pneumoniae, 100K- a 100500 frames MD of Cyclophilin A based on PDB 2N0T, and 250K- a 250000 frames MD of four chains of the Tau peptide that corresponds to the MD simulation of an extended Tau peptide (PHF8) during 1 microsecond.
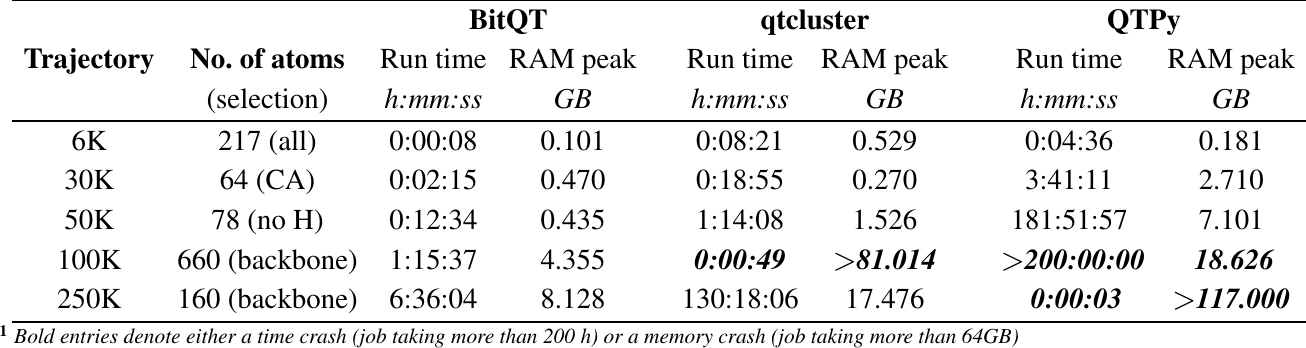
Figure 2: Performance benchmark of BitQT vs QTPy vs qtcluster.¶
All calculations were performed on an AMD Ryzen5 Hexa-core Workstation with a processor speed of 3.6 GHz and 64GB RAM under a 64-bit Xubuntu 18.04 operating system. Run times and RAM peaks were recorded with the emph{/usr/bin/time} Linux command.
For more details, please refer to the supporting information of the academic publication.
Useful Alternatives¶
As we have described, BitQT is an heuristic approach that can be used as a replacement for the very time-consuming exact variants of Quality Threshold clustering of Molecular Dynamics. However, there exist other cheaper, popular, useful alternatives for geometrical clustering that might equally fit your needs. Here you go …
QTPy is an exact implementation of the original Quality Thresold for Molecular Dynamics. Technically, this ones is not cheaper, but you might want to consider it for benchmark purposes. Implemented using an RMSD square matrix of floats (half-precision).
BitClust is an exact implementation of a very popular clustering algorithm. You may have heard of it as daura, qt-like, qt, neighbor-based or gromos. It has been implemented in VMD, GROMACS, WORDOM, PyPROCT and others. BitClust is implemented using an RMSD-encoded square matrix of bits.
RCDPeaks is, to the best of our knwoledge, the first exact implementation of Density Peaks clustering that does not need a square matrix of floats. Instead, it uses a dual-heap approach so it is very lightweight and faster than other alternatives.
MDSCAN is an alternative to HDBSCAN that uses RMSD as metric but does not need a square matrix to work as it was implemented using an efficient dual-heap approach. HDBSCAN is perhaps on of the most robust clustering algorithms out there. It has been succesfully apllied to Molecular Dynamics. However, most implementations do not include RMSD as similarity metric. The workaround for those alternatives is to accept a precomputed square float matrix that is too costly when dealing with long trajectories.